MEDT340N Study Guide - Midterm Guide: Petechia, Capillary, The X Factor

21 Oct 2013
School
Department
Course
Professor
Document Summary
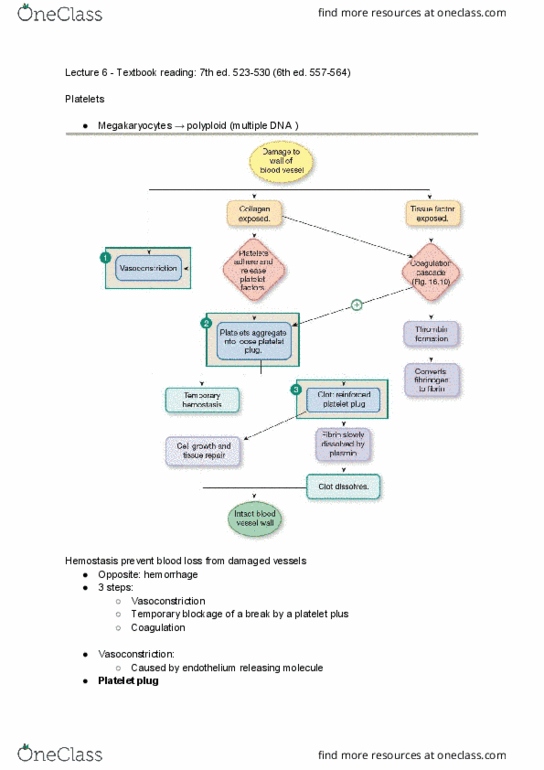
1 plug- not permanent until fibrin clot: platelets tested for, number- (150,000-450,000/ul, thrombocytopenia, abs destroy, decreased production in bone marrow. Is it functioning: glanzmanns- missing thrombasthenin, **testing for platelet number, thrombocytopenia would interfere with test, tourniquet test- rumple leede. In vivo: serial plt counts from finger stick or bleeding time stick, see how decreases over time, abnormal vascular function- no specific test, blood vessel and/or platelet function is problem. **tests all but iii, iv (ca++), vii, xiii (not plug waiting) and pf3 (b/c add substitute: thrombin time, normal= 15 seconds, **testing for i (fibrinogen, coagulation sequence, platelet aggregation, plasma coagulation factors and synonyms- Serum prothrombin conversion accelerator (spca); stabile prothrombin conversion factor; proconvertin. Cofactor for activation of prothrombin to thrombin w/ tissue thromboplasin, initiates the extrinsic pathway. Activated form is enzyme for intrinsic activation of factor x. Activated form is enzyme for final common pw activation of prothrombin. Activated form is intrinsic activator of factor ix.