BIOL266 Lecture Notes - Lecture 5: Dna Annotation, Blast, Reading Frame

25 Sep 2017
School
Department
Course
Professor
Document Summary
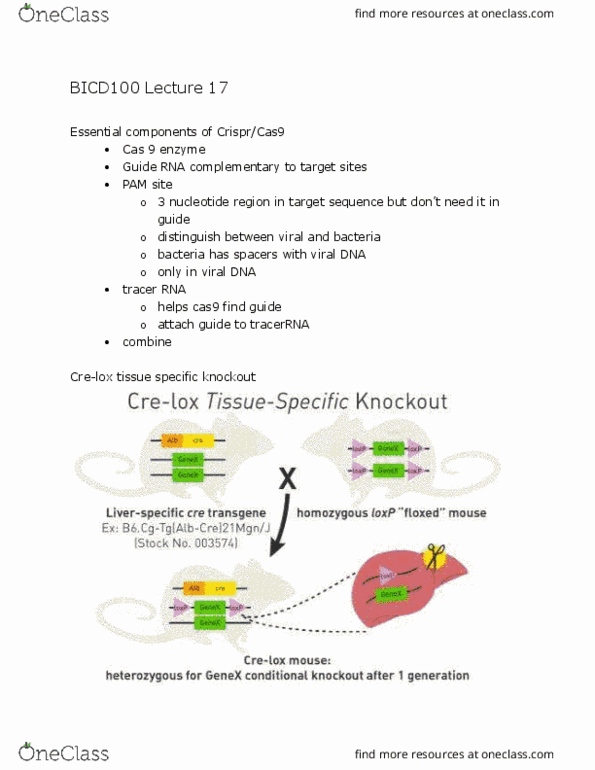
We now know how to align pairs of sequences (1 vs 1) Now suppose we have a sequence (query) and want to find its closest homologs in a database. To find closest db homolog of a given sequence. Repeat the alignment to all the databases. Suppose 3 and 5 are true homologs of the query sequence and all other sequences are unrelated. We need an algorithm to rapidly compare the query sequence against all target sequences in. Each target must be given a score reflecting degree of similarity. We then need to estimate the probability that the match could occur by chance. The score should reflect the degree of evolutionary similarity. If you can calculate the probability that the random good alignments happen then you can find the random bad alignment that occur. Instead of 1 vs 1 sw, perform 1 vs many. Very slow when searching large databases (e. g. , genbank) Gaining favour with increase in computer speed!