BIOC34H3 Lecture Notes - Lecture 5: Parasympathetic Nervous System, Bradycardia, Sympathetic Nervous System

Document Summary
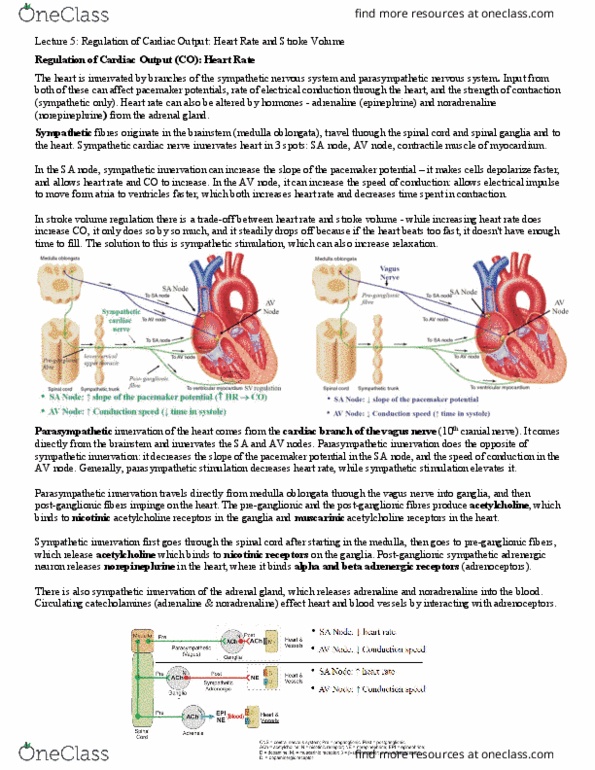
Lecture 5: regulation of cardiac output: heart rate and stroke volume. The effects of sympathetic regulation upon the heart rate can be rather complex. At the level of the pacemaker cells, noradrenaline (released from sympathetic nerves and the adrenal gland) and adrenaline (released from the adrenal gland) bind beta adrenergic receptors on the pacemaker cells. These, in turn, activate stimulatory g proteins, which go onto to activate adenylyl cyclase, to produce cyclic amp (camp). A phosphorylation cascade occurs, activating protein kinases, and these open funny channels, and t-type ca2+ channels. Na+, in the first phase of the pacemaker potential, and ca2+ in the second phase of the pacemaker potential, enter the cell rapidly (more so than in the absence of sympathetic stimulation) and trigger a quicker depolarization than normal. This means firing threshold is reached sooner and heart rate can increase.