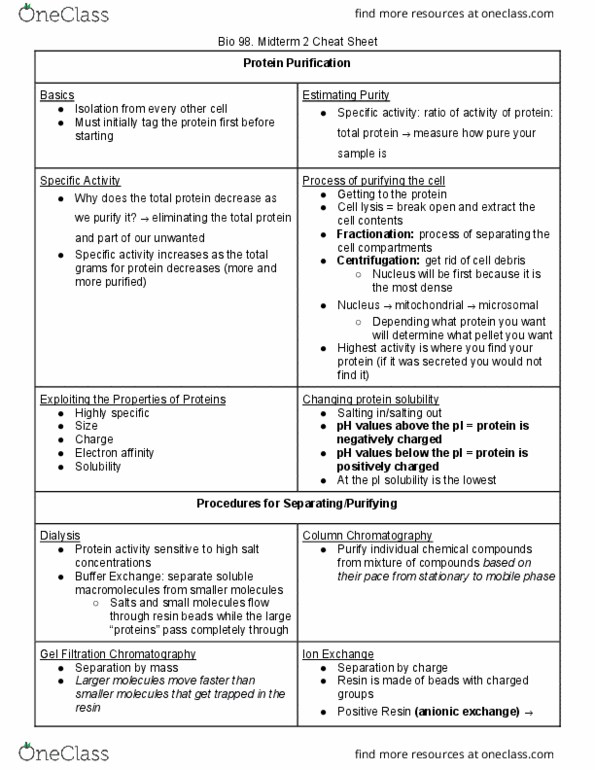
In class we expressed and purified the human form of LDH. Our purification protocol began with the Ni-affinity column. However, as you should have all noted, some significant containments still remained in our âpooledâ sample. To remove these impurities we could have gone on and ran other chromatography columns such as ion exchange, size-exclusion columns, and other affinity columns. Given this, along with the sequence of LDH below, answer the following questions: 1 MATLKDQLIY NLLKEEQTPQ NKITVVGVGA VGMACAISIL MKDLADELAL VDVIEDKLKG 61 EMMDLQHGSL FLRTPKIVSG KDYNVTANSK LVIITAGARQ QEGESRLNLV QRNVNIFKFI 121 IPNVVKYSPN CKLLIVSNPV DILTYVAWKI SGFPKNRVIG SGCNLDSARF RYLMGERLGV 181 HPLSCHGWVL GEHGDSSVPV WSGMNVAGVS LKTLHPDLGT DKDKEQWKEV HKQVVESAYE 241 VIKLKGYTSW AIGLSVADLA ESIMKNLRRV HPVSTMIKGL YGIKDDVFLS VPCILGQNGI 301 SDLVKVTLTS EEEARLKKSA DTLWGIQKEL QF*
a. What is the theoretical pI of human LDH? Given this pI would a Qsepharose column be a good choice for further purification? Why or why not?
b. Would you expect LDH to bind to a HiTrapTM Blue HP column? Why or why not?
c. A typical last step of a purification protocol involves running your protein sample over a size exclusion column. Letâs say we do this for LDH. At first you choose a column with a molecular weight cutoff of 80 kDa. Due to the size of your protein you expect it to come off somewhere in the middle of the run and foolishly decide to start collecting fractions halfway through the run. When you come back you see your data and realize the protein came off in the âvoidâ volume (very early in the run) suggesting that your protein is much larger than 80 kDa. What is a possible explanation for this data?
In class we expressed and purified the human form of LDH. Our purification protocol began with the Ni-affinity column. However, as you should have all noted, some significant containments still remained in our âpooledâ sample. To remove these impurities we could have gone on and ran other chromatography columns such as ion exchange, size-exclusion columns, and other affinity columns. Given this, along with the sequence of LDH below, answer the following questions: 1 MATLKDQLIY NLLKEEQTPQ NKITVVGVGA VGMACAISIL MKDLADELAL VDVIEDKLKG 61 EMMDLQHGSL FLRTPKIVSG KDYNVTANSK LVIITAGARQ QEGESRLNLV QRNVNIFKFI 121 IPNVVKYSPN CKLLIVSNPV DILTYVAWKI SGFPKNRVIG SGCNLDSARF RYLMGERLGV 181 HPLSCHGWVL GEHGDSSVPV WSGMNVAGVS LKTLHPDLGT DKDKEQWKEV HKQVVESAYE 241 VIKLKGYTSW AIGLSVADLA ESIMKNLRRV HPVSTMIKGL YGIKDDVFLS VPCILGQNGI 301 SDLVKVTLTS EEEARLKKSA DTLWGIQKEL QF*
a. What is the theoretical pI of human LDH? Given this pI would a Qsepharose column be a good choice for further purification? Why or why not?
b. Would you expect LDH to bind to a HiTrapTM Blue HP column? Why or why not?
c. A typical last step of a purification protocol involves running your protein sample over a size exclusion column. Letâs say we do this for LDH. At first you choose a column with a molecular weight cutoff of 80 kDa. Due to the size of your protein you expect it to come off somewhere in the middle of the run and foolishly decide to start collecting fractions halfway through the run. When you come back you see your data and realize the protein came off in the âvoidâ volume (very early in the run) suggesting that your protein is much larger than 80 kDa. What is a possible explanation for this data?